
Natalia Garrido, paciente PKU: "No sufro retraso mental porque mi doctora se encadenó a un árbol"
Natalia Garrido, paciente PKU: "No sufro retraso mental porque mi doctora se encadenó a un árbol"
En el Día Mundial de las Enfermedades Raras, Natalia Garrido nos explica qué es la fenilcetonuria y cómo influye en su día a día.
Noticias relacionadas
Natalia Garrido es una de las 1.500 personas que padecen fenilcetonuria en España, una enfermedad rara también conocida como PKU. Se trata de una alteración congénita del metabolismo, causada por la carencia de la enzima fenilalanina hidroxilasa que incapacita al hígado para metabolizar el aminoácido. “Para que se entienda mejor, no puedo comer proteínas y las proteínas están en casi toda la comida. Solo puedo comer ‘libre’ fruta y verdura”.
Garrido habla en el Día Mundial de las Enfermedades Raras (28 de febrero) sobre la importancia de investigar este tipo de enfermedades y realizar, desde edades tempranas, pruebas para detectarlas.
"El tiempo es vital para evitar el daño cognitivo"
“Me gustaría concienciar sobre la importancia de que los bebés se hagan la prueba de talón dentro de los 10 primeros días de nacimiento. El tiempo es vital para evitar el daño cognitivo. Cuanto antes se ponga a dieta al bebé, menos daño sufrirá su cerebro y más posibilidades tendrá de desarrollar una vida normal”.
A sus 23 años, Garrido lleva una vida normal, estudia un máster de Producción Audiovisual en la Escuela TAI y no sufre ningún trastorno cognitivo o motor.
Sin embargo, si ella o cualquier otro paciente fenilcetonúrico se saltara su estricta dieta, las proteínas que consumiera se convertirían en toxinas y afectarían al sistema nervioso central y al cerebro causando epilepsia, retraso motor, retraso mental…

¿Fenil… qué?
La fenilcetonuria es una enfermedad genética que se transmite por el ADN. Ambos padres deben ser portadores aunque no sufran la alteración y se detecta en la prueba de talón que se realiza a los recién nacidos.
La doctora Mercedes Martínez-Pardo es un referente, ha sido una de las pioneras en el tratamiento de la enfermedad en España. Durante muchos años se ha ocupado del seguimiento de la enfermedad cada vez que un recién nacido daba positivo en fenilcetonuria en la Comunidad de Madrid.
Los fenilcetonúricos le deben mucho. Fue quien consiguió que la medicina en España fuese subvencionada por la Seguridad Social. Tuvo que pelearlo. Ante la negativa gubernamental a dar la subvención, la doctora Martínez-Pardo se ató a un árbol frente al Ministerio de Sanidad.
"No sufro retraso mental porque mi doctora se encadenó a un árbol"
Cada caja de medicinas dura a los pacientes una semana y cuesta 750 euros. “Gasto unas cuatro cajas al mes, tendría que pagar alrededor de 3.000 euros mensuales si la medicina no estuviese subvencionada por la Seguridad Social. No sé si podría pagarlo, tampoco si el resto de familias con familiares fenilcetonúricos".
El tratamiento es un suplemento proteico, ya que los pacientes deben seguir una dieta baja en proteínas. El suplemento es una especie de batido de proteínas que contiene los aminoácidos que sí pueden tomar y son vitales para el desarrollo.
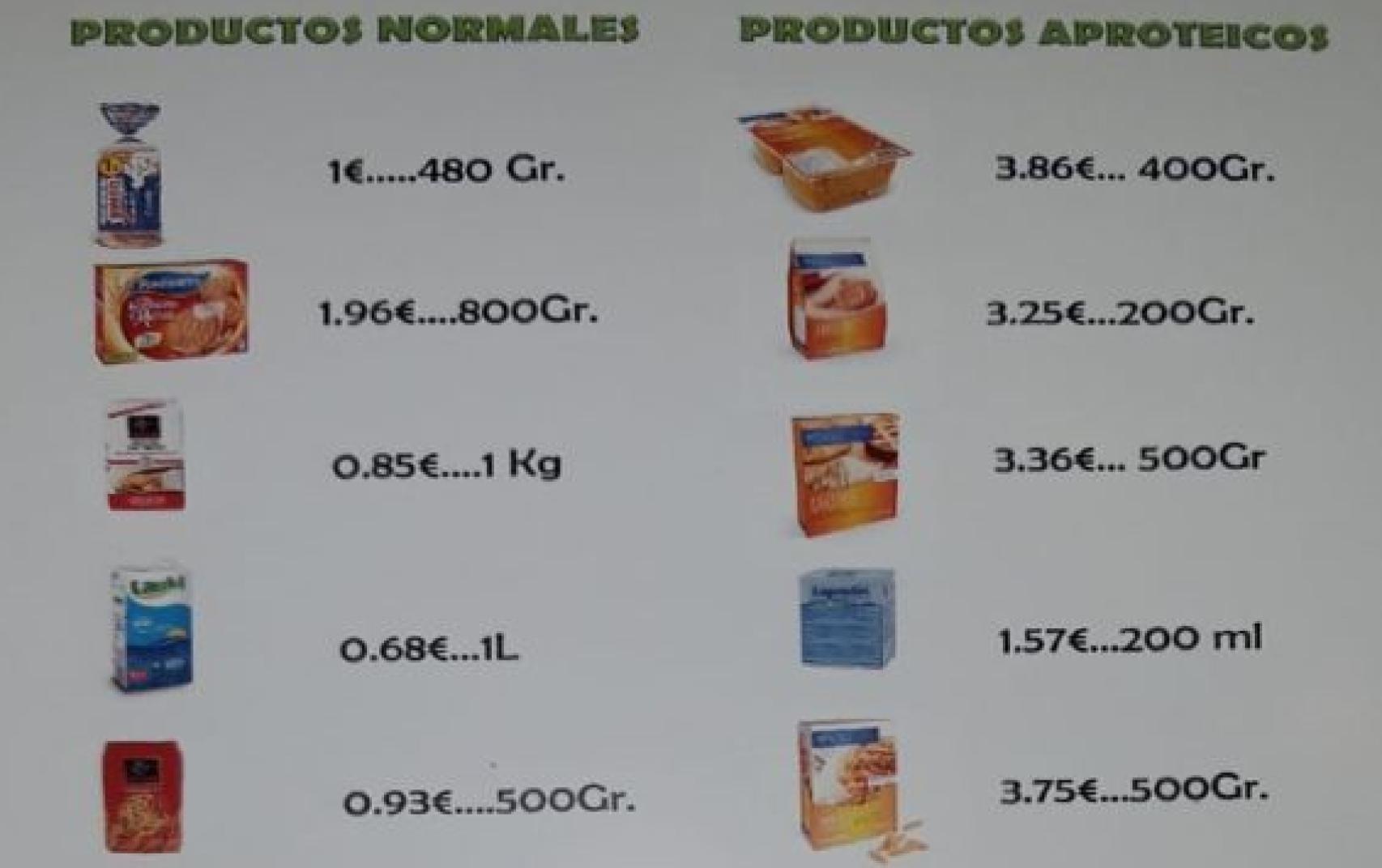
Comida 8 veces más cara
Hay alimentos bajos o sin proteínas que no se encuentran en supermercados, así que hay asociaciones y organizaciones que los suministran.
Una de ellas es ASFEMA, fundada por los padres de los primeros niños diagnosticados en Madrid, que forma parte de la Federación de Enfermedades Metabólicas de España. “Ellos se encargan de gestionar que llegue nuestra ‘comida especial’, pero también realizan otras actividades de apoyo a las familias”.
El problema es que estos alimentos no están subvencionados y cuestan ocho veces más que los que encontramos en los supermercados.
Por otra parte, hay que tener en cuenta que cada paciente tolera una cantidad de proteínas diarias y es importante que tome esa cuantía porque "es igual de malo pasarse por arriba que por abajo".
“En mi caso puedo comer, por ejemplo, dos huevos al día, o una tostada y un huevo, o un pequeño plato de pasta. La gente ‘normal’ puede tomar la cantidad que quiera de proteínas y su hígado se encarga de ‘filtrar’ la fenilalanina que necesita. Los PKU tenemos ese ‘filtro’ roto así que tenemos que hacer nosotros los cálculos”.
Pregunta: ¿Cómo influye la fenilcetonuria en tu día a día?
Respuesta: Tengo que tener cuidado al comer fuera, puedo optar por comer ensaladas, pequeños platos de pasta, patatas fritas… o llevarme mi propia comida. Cada vez que comes por primera vez con alguien o cambias de entorno y te ven el táper tienes que explicar todo de nuevo.
Una maleta para la comida
Además, a la hora de viajar llevo dos maletas, una con la ropa y otra con las medicinas y la comida.
Y, con la vista puesta a largo plazo, tendré que planificar el embarazo. Las mujeres con fenilcetonuria tenemos embarazos de riesgo, así que primero tenemos que restringir aún más nuestra dieta en proteínas y mantenerla así durante los nueve meses de embarazo para que el feto no sufra malformaciones.
¿Hay opciones de mejora?
Es una enfermedad crónica y, por tanto, la dieta es para toda la vida. Para algunos pacientes hay una nueva medicina que se toma en forma de pastillas, BH4 o Kuvan, con la que pueden llevar una dieta normal o casi normal. El problema es que no funciona en todos los pacientes, depende del nivel de tolerancia a la fenilalanina en sangre.
¿Ha influido la pandemia en el tratamiento?
Sí. Todo el dinero dedicado a la investigación está centrado en el coronavirus y el resto de enfermedades, sobre todo las raras, nos estamos quedando atrás.
En Estados Unidos van un paso por delante y los pacientes como yo se inyectan una sustancia, PegPal, que en el caso de funcionar, te permite llevar una dieta normal. La pandemia ha retrasado su llegada a España.
El descubrimiento
En 1934, el médico noruego Asbörjn Fölling descubrió la fenilcetonuria (PKU).
Se dio cuenta de que la orina de algunos pacientes con retraso mental severo olía “raro”. Entonces, el doctor pensó que les debía pasar algo relacionado con el metabolismo y así empezaron las investigaciones.
En 1980, investigadores de Boston descubrieron los problemas que tenían los niños con esta enfermedad no tratados. Entonces el National Institute of Child Health of Human Development empezó un estudio que demostró que la terapia dietética evitaba la discapacidad intelectual.



